Manual¶
This page contains the details on how to use the functions that DICEseq provides. Before using diceseq and dice-count, you need the annotation file, which you could download from sepcific database, e.g., Ensembl, but you may need to change it to the format that diceseq supports.
1. preprocess¶
1.1 annotation format¶
The basic format is gtf. In order to simply process the gtf file, we used a few formats of key words. The default is similar as Ensembl format. Namely, the order is gene –> transcript 1 –> exon ... –> transcript 2 –> exon ... , and the attributes includes gene_id, gene_name, ect. as follows
XV ensembl gene 93395 94402 . - . gene_id "YOL120C"; gene_name "RPL18A"; gene_biotype "protein_coding";
XV ensembl transcript 93395 94402 . - . gene_id "YOL120C"; gene_name "RPL18A"; gene_biotype "protein_coding";
XV ensembl exon 94291 94402 . - . gene_id "YOL120C"; gene_name "RPL18A"; gene_biotype "protein_coding";
XV ensembl exon 93395 93843 . - . gene_id "YOL120C"; gene_name "RPL18A"; gene_biotype "protein_coding";
XI ensembl gene 431906 432720 . + . gene_id "YKL006W"; gene_name "RPL14A"; gene_biotype "protein_coding";
XI ensembl transcript 431906 432720 . + . gene_id "YKL006W"; gene_name "RPL14A"; gene_biotype "protein_coding";
XI ensembl exon 431906 432034 . + . gene_id "YKL006W"; gene_name "RPL14A"; gene_biotype "protein_coding";
XI ensembl exon 432433 432720 . + . gene_id "YKL006W"; gene_name "RPL14A"; gene_biotype "protein_coding";
1.2 alignment¶
There are quite a fewer aligner that allows mapping reads to genome reference with big gaps, mainly caused by splicing. You could use STAR, TOPHAT, but I would suggest HISAT here, which is fast and returns reads with good aligment quality.
You could run it like this (based on HISAT 0.1.5), which including alignment, sort and index:
($hisatDir/hisat -x $hisatRef -1 $fq_dir/"$file"_1.fq.gz -2 $fq_dir/"$file"_2.fq.gz --no-unal | samtools view -bS -> $out_dir/$file.bam) 2> $out_dir/$file.err
samtools sort $out_dir/$file.bam $out_dir/$file.sorted
samtools index $out_dir/$file.sorted.bam
2. diceseq¶
This command allows you to estimate isoform proportions jointly (or separately if you only input one time point each time). It also allows to merging multiple replicates. You could run it like this:
my_sam_list=t1_rep1.sorted.bam,t1_rep2.sorted.bam---t1_rep1.sorted.bam---t3_rep1.sorted.bam
diceseq -a anno_file.gtf -s $my_sam_list -t 1,2,5 -o out_file
There are more parameters for setting (diceseq -h always give the version you are using):
Usage: diceseq [options]
Options:
-h, --help show this help message and exit
-a ANNO_FILE, --anno_file=ANNO_FILE
Annotation file for genes and transcripts in GTF or
GFF3
-s SAM_LIST, --sam_list=SAM_LIST
Sorted and indexed bam/sam files, use ',' for
replicates and '---' for time points, e.g.,
T1_rep1.bam,T1_rep2.bam---T2.bam
-t TIME_SEQ, --time_seq=TIME_SEQ
The time for the input samples [Default: 0,1,2,...]
-o OUT_FILE, --out_file=OUT_FILE
Prefix of the output files with full path
Optional arguments:
-p NPROC, --nproc=NPROC
Number of subprocesses [default: 4]
--add_premRNA Add the pre-mRNA as a transcript
--fLen=FRAG_LENG Two arguments for fragment length: mean and standard
diveation, default: auto-detected
--bias=BIAS_ARGS Three argments for bias correction:
BIAS_MODE,REF_FILE,BIAS_FILE(s). BIAS_MODE: unif,
end5, end3, both. REF_FILE: the genome reference file
in fasta format. BIAS_FILE(s): bias files from dice-
bias, use '---' for time specific files, [default:
unif None None]
--thetas=THETAS Two arguments for hyperparameters in GP model:
theta1,theta2. default: [3 None], where theta2 covers
1/3 duration.
--mcmc=MCMC_RUN Four arguments for in MCMC iterations:
save_sample,max_run,min_run,gap_run. Required:
save_sample =< 3/4*mim_run. [default: 0 20000 1000
100]
Suggestions on setting hyperparameter 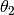 : if you want cover
: if you want cover 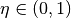 of duration, then you should set
of duration, then you should set 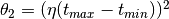 . The default is
. The default is 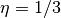 . Generally, we suggest using a small , e.g., covering less than 1/3 length, while it really depends on the time scale.
. Generally, we suggest using a small , e.g., covering less than 1/3 length, while it really depends on the time scale.
For output, you may see two files out_file.dice and out_file.sample.gz. As the default of sample_num=0, you won’t see the xx.sample.gz file. An example of xx.dice file is like this.
tran_id gene_id logLik transLen FPKM_T0 ratio_T0 ratio_lo_T0 ratio_hi_T0 FPKM_T1 ratio_T1 ratio_lo_T1 ratio_hi_T1 FPKM_T2 ratio_T2 ratio_lo_T2 ratio_hi_T2
YMR116C.m YMR116C -3.8e+03 960 3.80e+04 0.472 0.366 0.577 6.30e+04 0.680 0.595 0.757 9.38e+04 0.885 0.837 0.940
YMR116C.p YMR116C -3.8e+03 1233 4.25e+04 0.528 0.423 0.634 2.97e+04 0.320 0.247 0.405 1.21e+04 0.115 0.060 0.164
YKL006W.m YKL006W -2.1e+03 417 2.36e+04 0.292 0.195 0.393 5.34e+04 0.583 0.471 0.683 9.00e+04 0.850 0.769 0.925
YKL006W.p YKL006W -2.1e+03 815 5.70e+04 0.708 0.608 0.805 3.83e+04 0.417 0.318 0.529 1.58e+04 0.150 0.075 0.233
It contains the 4+4t columns, with 4 basic features and 4 features for each time:
- column 1: transcript id
- column 2: gene id
- column 3: log likelihood at gene level
- column 4: transcript length
- column 5: FPKM for a time point
- column 6: isoform fraction for a time point
- column 7: lower bound of 95% confidence interval of isoform fraction
- column 8: higher bound of 95% confidence interval of isoform fraction
3. dice-count¶
This command allows you to calculate the reads counts in an aligned + sorted + indexed sam (or bam) file with an annotation file in gtf format. It allows calculating the total counts for each gene, but also specific counts of different segments (e.g., junction, exon, and intron) if a gene has exactly one intron. You could run it like this:
dice-count -a anno_file.gtf -s sam_file.bam -o out_file.txt
There are more parameters for setting (dice-count -h always give the version you are using):
Usage: dice-count [options]
Options:
-h, --help show this help message and exit
-a ANNO_FILE, --anno_file=ANNO_FILE
Annotation file for genes and transcripts
-s SAM_FILE, --sam_file=SAM_FILE
Sorted and indexed bam/sam files
-o OUT_FILE, --out_file=OUT_FILE
The counts in tsv file
Optional arguments:
-p NPROC, --nproc=NPROC
Number of subprocesses [default: 4]
--anno_type=ANNO_TYPE
Type of annotation file: GTF, GFF3, UCSC_table
[default: GTF]
--mapq_min=MAPQ_MIN
Minimum mapq for reads. [default: 10]
--mismatch_max=MISMATCH_MAX
Maximum mismatch for reads. [default: 5]
--rlen_min=RLEN_MIN
Minimum length for reads. [default: 1]
--overhang=OVERHANG
Minimum overhang on junctions. [default: 1]
--duplicate keep duplicate reads; otherwise not
--partial keep reads partial in the region; otherwise not
--single_end use reads as single-end; otherwise paired-end
--junction return junction and boundary reads, only for gene with
one exon-intron-exon structure; otherwise no junction.
An output without --junction:
gene_id gene_name biotype gene_length count FPKM
YMR116C ASC1 protein_coding 1233 100 8.53e+04
YKL006W RPL14A protein_coding 815 43 5.55e+04
YNL112W DBP2 protein_coding 2643 179 7.12e+04
Another output with --junction:
gene_id gene_name biotype gene_length ex1_NUM ex1_int_NUM int_NUM int_ex2_NUM ex2_NUM ex1_ex2_junc_NUM ex1_int_ex2_NUM ex1_ex2_vague_NUM ex1_FPKM ex1_int_FPKM int_FPKM int_ex2_FPKM ex2_FPKM ex1_ex2_junc_FPKM ex1_int_ex2_FPKM ex1_ex2_vague_FPKM
YKL006W RPL14A protein_coding 815 0 4 2 5 14 9 1 8 0.00e+00 3.26e+01 1.05e+01 2.54e+01 1.80e+02 7.33e+01 8.14e+00 1.53e+02
YOL120C RPL18A protein_coding 1008 0 2 7 4 38 7 1 5 0.00e+00 1.88e+01 2.87e+01 2.20e+01 1.64e+02 6.57e+01 9.38e+00 1.38e+02
YMR116C ASC1 protein_coding 1233 36 6 2 1 26 22 1 6 1.10e+02 3.09e+01 2.96e+01 4.45e+00 1.23e+02 6.64e+01 2.48e+00 1.81e+01
Both return reads count and FPKM. For the junction output, it contains reads in 8 regions (in the way of one-intron gene, for exon skipping the order event, change accordingly):
- within exon 1
- boundary of exon 1 and intron
- within intron
- boundary of intron and exon 2
- within exon 2
- junction between exon 1 and exon 2
- overlap of all exon 1, intron and exon 2
- unsure when one mate in exon 1 and the other mate in exon 2
This option gives the option to have junction and boundary reads, but only desinged for one-intron RNA splicing (in yeast) or exon skipping triplets.